Die Registrierung Ihrer Einrichtung als SoHO-Einrichtung (gemäß neuer EU-Verordnung Source of Human Origin) beginnt im September. Wir werden Sie hier informieren!
Podiumsdiskussion mit Proffs. A. Steinbicker, Köln; D. Fischer, Heidelberg; P. Meybohm, Würzburg; C. von Heymann, Berlin, K. Zacharowski, Frankfurt; J. Meier, Graz, J. Adili, Darmstadt.
Podiumsdiskussion mit Proffs. A. Steinbicker, Köln; D. Fischer, Heidelberg; M. Funk, Langen; K. Zacharowski, Frankfurt; F. Adidli, Darmstadt; M. Caspers, Witten/Herdecke
Es ist soweit - die Vorträge der diesjährigen 23. IAKH Transfusionsgespräche stehen für interessierte und registrierte Mitglieder zum Download bereit!!
Loggen Sie sich einfach in den Mitgliederbereich mit ihrem Passwort ein und folgen Sie diesem Link.
Auf Initiative der IAKH haben die Sektionen der DIVI-Sektionen „Klinische Hämotherapie und Hämostasemanagement“ und „Trauma“ unter Fries und Hamsen in einem Positionspapier den Stand der Wissenschaft zur prähospitalen Gabe allogener Blutprodukte zusammengefasst ...
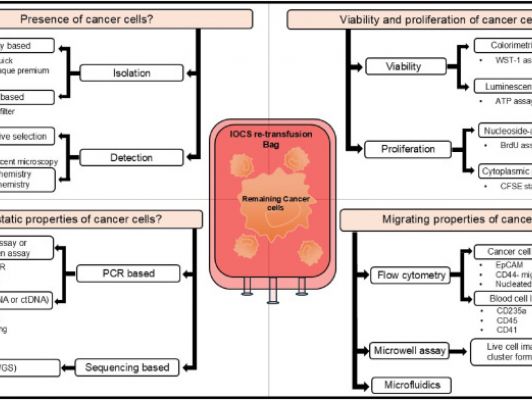
Die Erstautorin H. Sureshkumar und Koautoren aus Heidelberg haben in einem sehr guten Methodikartikel den derzeitigen Forschungsstand zum Nachweis von malignen Zellen im Retransfundat beim Einsatz einer maschinellen Autotransfusion (MAT) dargestellt...
Das mittlerweile verpflichtende Melderegister hat seit Jahrzehnten eine flächendeckende und aussagekräfte Analyse der Anwendung von Blutprodukten etabliert und ist das größte funktionierende Hämovigilanzsystem. Was hat sich neues Lernpotenzial ergeben? Lesen Sie die Kurzzusammenfassung...